Methoden der molekularen Evolutionsforschung
Man kann zunächst Methoden der molekularen Evolutionsforschung unterscheiden, die an Proteinen, und solche, die an Nucleinsäure ansetzen.
Die exaktesten Ergebnisse liefern in beiden Fällen Sequenzierungen. Andere Methoden bedienen sich ausgewählter homologer DNA-Abschnitte verschiedener Organismen. Je nach der Auswahl können dabei phylogenetische oder populationsgenetische Aussagen gemacht werden. Manche Methoden – z. B. der Genetische Fingerabdruck oder die Mikrosatelliten-PCR-Analyse – sind auch geeignet, um Unterschiede zwischen einzelnen Individuen herauszufinden (z. B. für Vaterschaftsnachweise oder in der Kriminalistik)
Methoden, die an Proteinen ansetzen
Aminosäuresequenzierung
Die Methode wurde vor allem zur Erforschung entfernter Verwandtschaften eingesetzt. Untersucht werden Enzyme wie Cytochrome, die bei fast allen Lebewesen vorkommen und die Aufeinanderfolge kodierter Aminosäuren im Erbmaterial.
Alloenzymanalyse
Unter Alloenzymen versteht man Enzyme, die von einem Genort (Locus) kodiert werden, für den es mehrere Allele gibt. Damit sind Alloenzyme ein Spezialfall der sogenannten Isoenzyme, worunter man Enzyme von gleicher oder fast gleicher Substrat- oder Wirkungsspezifität versteht, die jedoch in den Primärstrukturen mehr oder weniger große Unterschiede aufweisen.
Mit Hilfe der Alloenzymanalyse kann geprüft werden, ob Proteine eines Locus in ihren Aminosäuresequenzen identisch sind oder ob Punktmutationen in einem der Allele vorkommen, die man durch Elektrophorese (Bewegung geladener Moleküle einer Lösung im elektrischen Feld, d.h. mittels E. trennt man Moleküle einer Mischung z. B. aufgrund ihrer unterschiedlicher Ladung, Größe oder Struktur auf) trennen kann. Allerdings reichen die Trennbedingungen der Elektrophorese nicht immer aus, um diese sogenannten Elektromorphen (molekulare Bestandteile einer Mischung) tatsächlich zu trennen. Deshalb können mit dieser Methode nicht alle Varianten erfasst werden.
Zur Untersuchung werden in der Regel Markerproteine (Enzyme) verwendet, deren Aktivität man durch chromogene (Farbstoff bildende) Substrate visuell verfolgen kann wie z. B. Alkoholdehydrogenase, Adenylatkinase, Asparattransaminase, 6-Phosphatdehydrogenase, Hexokinase, Peptidase, Phosphoglucoseisomerase, Triosephosphatisomerase oder Xanthindehydrogenase. Die Untersuchung von Alloenzymen dient vor allem der populationsgenetischen Forschung an nah verwandten Arten, Unterarten oder Populationsgruppen. Junge und von wenigen Individuen gegründete Populationen zeigen im allgemeinen verminderten Polymorphismus (Vielgestaltigkeit), da sie nur einen Teil der Allelvielfalt der Elterngeneration mitnimmt. Ebenso begrenzten Polymorphismus zeigen Populationen, deren Größe einmal reduziert wurde und dann wieder anstieg. Es dauert oft über 1000 Generationen, bis die Heterozygotie (Mischerbigkeit) wieder ansteigt und den mittleren Wert großer Populationen erreicht (Gründereffekt und Flaschenhalseffekt). Ein solcher Gründereffekt ist z. B. für die in Europa eingebürgerten Waschbären, ein Flaschenhalseffekt für südafrikanische Palmfarne der Gattung Encephalartos nachgewiesen. Aus Alloenzymdaten lassen sich auch evolutionäre oder genetische Distanzen nach einem standardisierten Verfahren abschätzen (standard genetic distance).
Methoden, die an Nucleinsäuren ansetzen
DNA-DNA-Hybridisierung
Bei dieser Methode vereinigt (hybridisiert) man singel-copy-DNA (komplementäre Einzelstränge) von zwei verschiedenen Organismen. Nun kann man feststellen, um wie viel sich die Schmelztemperatur der Hybrid-DNA gegenüber der reinen DNA eines Organismus erniedrigt hat. Eine Verringerung der Schmelztemperatur um 1° C entspricht in etwa einem Unterschied von 1 %, d.h. dem Austausch eines Nucleotids in 100 Nucleotiden. Die Methode wurde 1966 von ELLIOT BRITTEN und D. KOHNE entwickelt. Mit dieser Methode können lediglich Ähnlichkeiten zwischen Organismen festgestellt werden, was eine Aufstellung von exakten Stammbäumen verhindert.
Restriktions-Fragment-Längenpolymorphismus (RFLP, restriction fragment length polymorphism)
Die Methode wurde vor allem in den Jahren 1970 bis 1990 für Mitochondrien-DNA (mtDNA) und Chloroplasten-DNA (cpDNA) angewandt. Sie wurde durch die Entdeckung von Ristriktionsendonucleasen durch WERNER ARBER im Jahre 1962 möglich. Mit Hilfe dieser Endonucleasen können DNA-Moleküle in Abschnitte definierter Länge zerschnitten werden, da diese Enzyme spezifische Sequenzabschnitte erkennen können, die häufig eine Palindromstruktur aufweisen (z. B. -G-A-A-T-T-C- Abb. 1). Auf diese Weise wurde z. B. die mitochondriale DNA von verschiedenen rezenten (noch bestehenden) Menschengruppen untersucht, was eine wesentliche Stütze für die „Out of Africa-Hypothese” lieferte. Die These besagt, dass sich der moderne Mensch (Homo sapiens sapiens) in Afrika entwickelte, von da ausgehend die restlichen Kontinente besiedelte und dort die anderen Hominiden (Aufrechtgehende) nach und nach verdrängte.
RAPD-PCR-Analyse
Voraussetzung für diese Methode war die Entwicklung der Polymerasekettenreaktion (PCR) in den Jahren 1985-1985 durch KAY MULLIS und seine Mitarbeiter. Mithilfe einer hitzeresistenten DNA-Polymerase (z. B. Taq-Polymerase aus dem thermophilen Bakterium Thermus aquaticus) kann man DNA-Abschnitte durch Erwärmen und leichtes Abkühlen und Wiedererwärmen in kurzer Zeit um den Faktor vermehren. Damit kann man aus winzigen DNA-Spuren soviel DNA produzieren, dass man eine anschließende Sequenzierung dieser DNA durchführen kann. Bei der RAPD-Analyse (randomly amplified polymorphic DNA) wird die Gesamt-DNA von unterschiedlichen Individuen mit einem Zufallsprimer von 10 Nucleotiden Länge mit Hilfe von PCR vervielfacht. Die erhaltenen Produkte werden durch Agarose-Gelelektrophorese aufgetrennt und durch Färbung sichtbar gemacht. Das unterschiedliche Bandenmuster ist ein Maß für die Unterschiede in den DNA-Abschnitten, d.h. für unterschiedliche Genloci bzw. Allele. Da diese Methode sich nicht immer als einwandfrei reproduzierbar herausstellte, spielt sie heute nur noch eine geringe Rolle.
-
RAPD-PCR
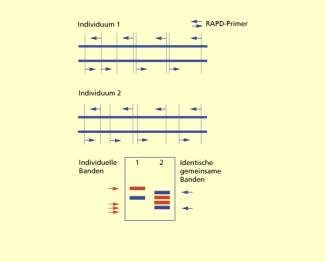

Mikrosatelliten-PCR-Analyse
Bei dieser Methode werden PCR-Primer (Hilfselemente, an dem die DNA-Polymerase den Replikationsvorgang beginnen kann, werden später abgebaut und durch DNA-Sequenzen ersetzt) von etwa 20 Nucleotiden Länge verwendet. Die Methode basiert auf dem Vorkommen kurzer, tandemartig angeordneter repetitiver (sich wiederholender) DNA-Sequenzen, die über das gesamte Genom eines Organismus verteilt sind (Mikrosatelliten-, Satelliten-DNA, repetitive DNA). Sie unterliegen innerhalb einer Population in der Anzahl ihrer Repetitionen und damit in ihrer Länge sehr starken Schwankungen. Dabei können Deletionen, Duplikationen oder auch Insertionen anderer Wiederholungseinheiten eine Rolle spielen (VNTR: variabel number of tandem repeats). Bei der Mikrosatelliten-PCR werden PCR-Primer eingesetzt, die spezifische Mikrosatellitengruppen begleiten. Die Länge der erhaltenen PCR-Produkte hängt von der Länge, d.h. der Gesamtzahl der wiederholten Mikrosatelliten-Elemente, ab. Von jeder doppelsträngigen DNA eines Individuums erhält man bei der Gelelektrophorese zwei Banden, die jeweils einem väterlichen und einem mütterlichen Allel entsprechen. Auf diese Art und Weise kann man, wenn man mehrere verschiedene Mikrosatelliten-Loci verwendet, sehr effektive Vaterschaftsnachweise durchführen. In der Evolutionsforschung ist diese Methode nur für populationsgenetische Untersuchungen unterhalb des Artniveaus geeignet.
DNA-Fingerprinting (genetischer Fingerabdruck)
Das DNA-Fingerprinting wurde 1985 von ALEC JEFFREYS entwickelt. Bei diesem Verfahren wird die Gesamt-DNA eines Individuums mit einer spezifischen Restriktionsendonuclease in unterschiedlich große aber definierte Restriktionsfragmente zerschnitten, die über Gelelektrophorese ihrer Größe nach aufgetrennt werden. Nach einem von SOUTHERN entwickelten Verfahren wird die aufgetrennte DNA dann auf eine Nylon- oder Nitrozellulosefolie übertragen. Nach ihrem Erfinder wird dieses Verfahren auch Southern-Blotting (engl. Blot = Fließpapier) genannt. Anschließend hybridisiert man ausgewählte DNA-Stücke mit markierten Sonden. Diese DNA-Sonden können entweder aus kurzen Oligonucleotiden (z. B. GGAT oder CAC) oder aus komplexen Mikrosatellitensequenzen bestehen. Nur wenn ausreichend viele Komplementärbasenpaare ausgebildet werden können, kommt eine sichere Hybridisierung zustande. Wurden die Sonden radioaktiv markiert kann man die DNA-Fragmente, an denen eine Sonde gebunden hat, durch Autoradiografie nachweisen. Dies führt zu den charakteristischen Bandenmustern.
Amplified fragment length polymorphism (AFLP-PCR)
Mit dieser Methode werden durch hochauflösende Gelelektrophorese Bandenmuster erzeugt, die Aufschluss über Variabilität zwischen Individuen, Populationen oder Arten geben. Die Methode wird vor allem für Genkartierungen bei Pflanzen eingesetzt. Sie eignet sich aber auch für Vaterschaftsanalysen.
Analyse von Nucleotid-Sequenzen
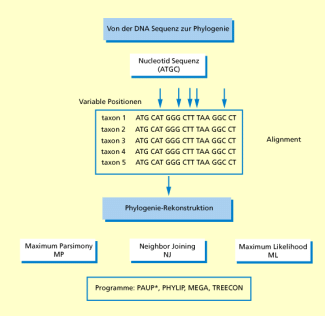
Die Analyse von Nucleotid-Sequenzen und eventuell auch von Aminosäuresequenzen der Genprodukte ist für die molekulare Evolutionsforschung und für die Rekonstruktion von Stammbäumen besonders gut geeignet. Im Gegensatz zu den anderen genannten Methoden sind die gewonnenen Daten eindeutig (in Banden können sich immer verschiedene Moleküle aufhalten). Sie sind quantifizierbar und statistischen Tests zugänglich. Sie weisen einen hohen Informationsgehalt auf, jede Nucleotidposition kann als ein unabhängiges Einzelmerkmal gewertet werden. Das bedeutet, bei Sequenzierung nur eines Markergens, erhält man auf diese Weise 1000 und mehr Merkmale als Resultat. Man kann diese Ergebnisse über weite systematische Bereiche vergleichen, da homologe Gene direkt erkennbar sind und - wie moderne Forschungen zeigen - oft in weiten Bereichen unterschiedlichster Verwandtschaftsgruppen vorkommen. Jedes Nucleotid lässt sich einem bestimmten Ort auf dem Gen zuordnen, was wichtig für Homologieüberlegungen ist, und man kann auch mit diesen Analysen arbeiten, wenn keine Mutationen vorliegen.
-
Prinzip der Vaterschaftsanalyse über Mikrosatelliten-PCR
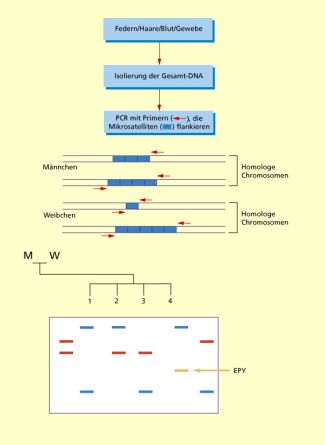

Bei dem Verfahren geht man so vor, dass man zunächst die Gesamt-DNA eines Organismus isoliert und dann ein Markergen auswählt, das durch Klonierung oder meistens durch PCR in solchen Mengen vermehrt wird, dass es anschließend sequenziert werden kann. In der Regel wählt man die Markergene aus nicht kodierender DNA oder aus Mitochondrien-DNA (bei Tieren) bzw. Chloroplasten-DNA (bei Pflanzen) aus. Diese Gene haben nichts mit der Morphologie zu tun und unterliegen deshalb kaum adaptiven Faktoren, welche die äußere Gestalt der Organismen verändert haben. Für weit zurückliegende evolutionäre Ereignisse und damit für die großen Zusammenhänge im Stammbaum eignen sich besonders die für die Ribosomen verantwortlichen Gene des Kerns, da sie in allen Eukaryoten gleichermaßen zur Verfügung stehen und sehr konservativ sind. rDNA-Gene in tierischen Mitochondrien sind wesentlich variabler und damit gute Marker für Ereignisse, die in den letzten 100 Millionen Jahren stattfanden. Bei dem Vergleich von Genen unterschiedlicher Organismen gibt es ein Problem. Neben sogenannten orthologen Genen, die wirklich homolog sind, gibt es Gene, die durch multiple aber unterschiedliche Kopien zu sogenannten Multigenfamilien geworden sind. Gene einer Multigenfamilie nennt man auch paralog. Es besteht die Gefahr, an Stelle homologer Gene solche paralogen Gene zu vergleichen, woraus dann natürlicherweise falsche Stammbäume entstehen.
Zur Genanalyse stehen folgende Methoden zur Verfügung:
- Genetische Kartierung (Kopplungsanalyse). Zunächst werden mit genetischen Markern, also bekannten DNA-Abschnitten, komplementäre Nucleotidsequenzen auf einem Chromosom markiert. Über die Analyse von Kopplungsgruppen mit anderen Nucleotidabschnitten einschließlich in ihrer Wirkung bekannter Gene kann so allmählich eine dichtere Karte der Nucleotidsequenzen gewonnen werden.
- Physische Kartierung. Ein Chromosom wird durch geeignete Restriktionsenzyme in eine überschaubare Zahl von Fragmenten zerlegt und dann wird deren Anordnung auf dem Chromosom bestimmt. Eine als Chromosome walking (Chromosomenwandern) bezeichnete Methode vereinigt verschiedene Techniken: Damit können Chromosomenkartierungen sehr beschleunigt werden.
-
Von der DNA-Sequenz zur Phylogenie

Suche nach passenden Schlagwörtern
- AFLP-PCR
- Out of Africa-Hypothese
- Multigenfamilie
- Flaschenhalseffekt
- PCR-Primer
- TAQ-Polymerase
- Stammbaum
- DNA-Fingerprinting
- Genanalyse
- Isoenzyme
- RAPD-PCR-Analyse
- Allele
- Aminosäuresequenz
- Mikrosatelliten-PCR-Analyse
- Nucleotidsequenz
- Evolution
- Restriktionsenzyme
- Genort
- Restriktionsfragment-Längenpolymorphismus
- Proteine
- Thermus aquaticus
- Polymerase-Kettenreaktion
- Markerproteine
- Klonierung
- Polymorphismus
- Markergene
- Locus
- RFLP-Analyse
- Alloenzyme
- Vaterschaftsnachweis
- Analyse von Nucleotid-Sequenzen
- genetischer Fingerabdruck
- Gründereffekt
- Alloenzymanalyse
- Antikörper
- Cladistik
- thermophile Bakterien

